Access the HPC (using MobaXterm on Windows or Terminal on Mac/Linux).
Explore raw FASTQ read files and understand the structure of FASTQ data.
Install new tools, set environment variables, or load pre-installed tools on the HPC system.
Check the quality of FASTQ reads using tools like FASTQC and interpret the output statistics.
Trim FASTQ reads to remove adapters or low-quality bases using appropriate tools.
Write a script (bash/python, etc.) to automate workflows, such as running commands on multiple files in a for loop, and submit jobs to an HPC system using SLURM SBATCH scheduling.
Tools to be Used
FASTQC / MULTIQC: For quality control of FASTQ reads.
Trimmomatic / fastp: For read trimming.
BWA: For mapping reads to a reference genome.
SAMTOOLS: For conversion of SAM to BAM files.
QUALIMAP: For quality control of BAM files.
Connecting to HPC (RAMSES) and Organizing the Workspace
Connect to the HPC
For Windows users: Use MobaXterm. For Mac/Linux users: Use the Terminal application.
Log in to the HPC cluster using your login credentials.
ssh-Y qbius030@@ramses1.itcc.uni-koeln.de# Replace 'qbius030' with your specific username.# Enter passphrase for key '/Users/tahirali/.ssh/ramses_key', if prompted:# Duo two-factor login for qbius030
Check Your Current Directory
pwd
If you are not in your home directory, navigate to it:
cd /home/group.kurse/qbius030/# Replace 'qbius030' with your specific username.
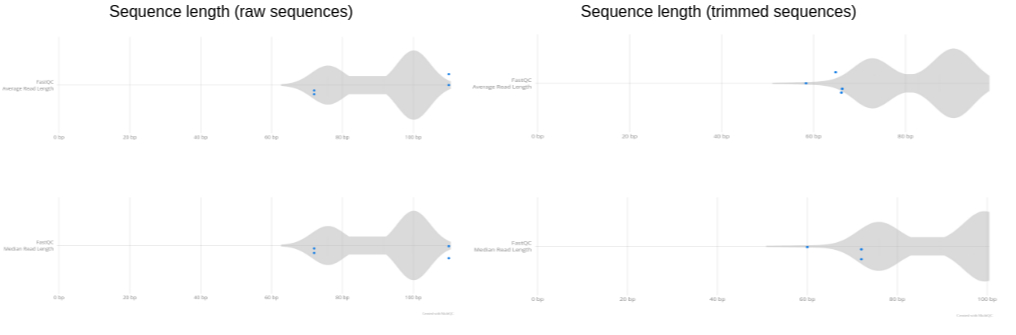
Compare Raw vs. Trimmed Reads with FastQC and MultiQC
Figure: plot showing read length distribution before and after trimming. ————————————————————————
Mapping to reference using BWA + SAMtools
Check if required module is installed:
module avail
Example SLURM script for Mapping:
#!/bin/bash -l#SBATCH --job-name=mapping#SBATCH --nodes=1#SBATCH --ntasks-per-node=8#SBATCH --mem=10gb#SBATCH --time=01:00:00#SBATCH --account=UniKoeln#SBATCH --output=/home/group.kurse/qbius030/Practical_Day_1/mapping/bwa_mapping.out#SBATCH --error=/home/group.kurse/qbius030/Practical_Day_1/mapping/bwa_mapping.errmodule purgemodule load bio/BWA/0.7.18-GCCcore-12.3.0module load bio/SAMtools/1.18-GCC-12.3.0INPUT_DIR="/home/group.kurse/qbius030/Practical_Day_1/trimming"OUTPUT_DIR="/home/group.kurse/qbius030/Practical_Day_1/mapping"REFERENCE="/home/group.kurse/qbius030/sample_project_data/reference_data/Arabidopsis_thaliana.TAIR10.dna.chromosome.4_98K.fasta"# bwa index $REFERENCE # Uncomment if not already indexedfor FORWARD_FILE in${INPUT_DIR}/*_paired_1.fastq.gz;doREVERSE_FILE="${FORWARD_FILE/_paired_1.fastq.gz/_paired_2.fastq.gz}"BASE_NAME=$(basename$FORWARD_FILE _paired_1.fastq.gz)SAM_FILE="${OUTPUT_DIR}/${BASE_NAME}.sam"BAM_FILE="${OUTPUT_DIR}/${BASE_NAME}.bam"SORTED_BAM="${OUTPUT_DIR}/${BASE_NAME}.sorted.bam"bwa mem -M-t 20 $REFERENCE$FORWARD_FILE$REVERSE_FILE>$SAM_FILEsamtools view -bS$SAM_FILE>$BAM_FILEsamtools sort -@ 20 $BAM_FILE-o$SORTED_BAMsamtools index $SORTED_BAMrm$SAM_FILErm$BAM_FILEdone
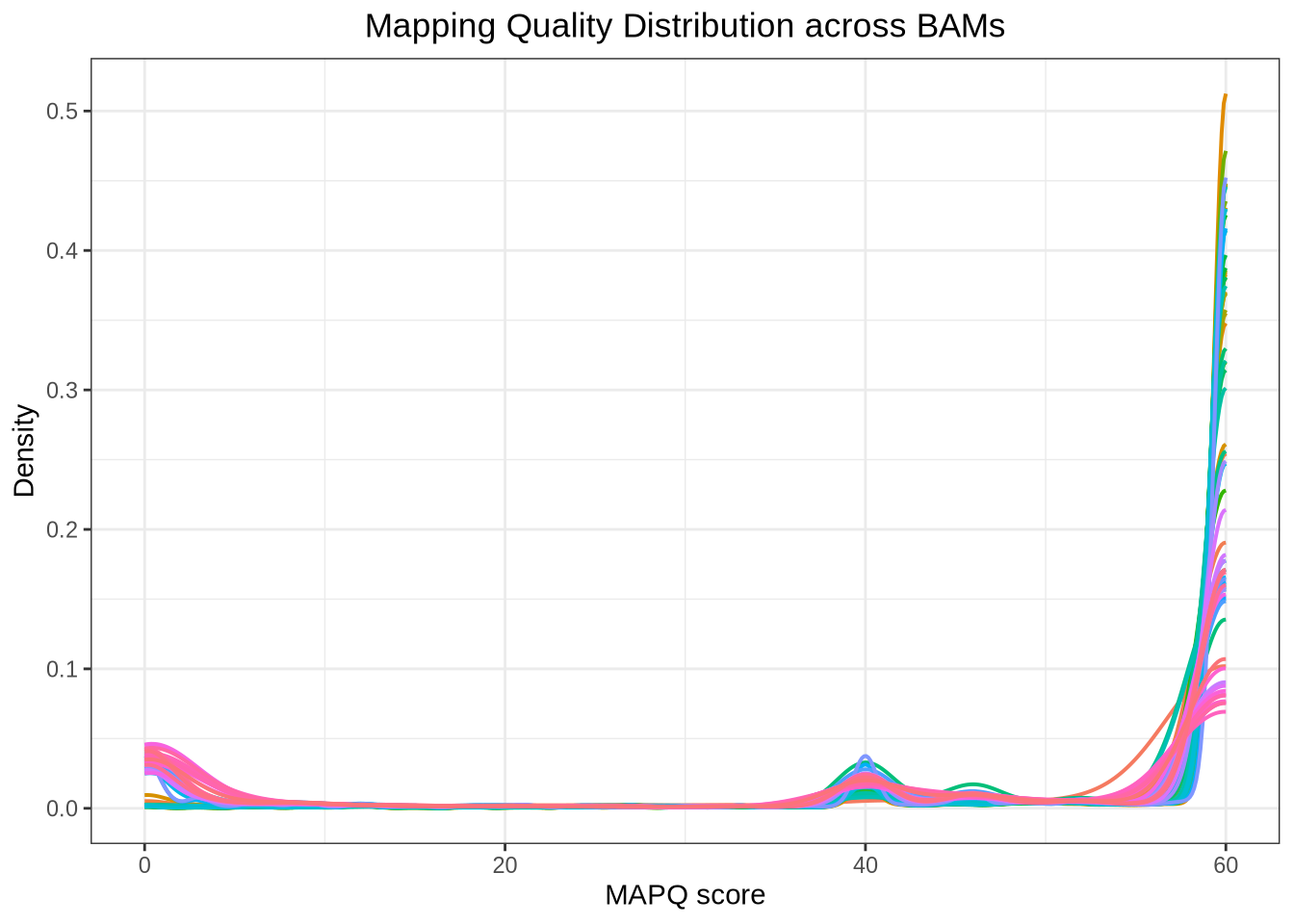
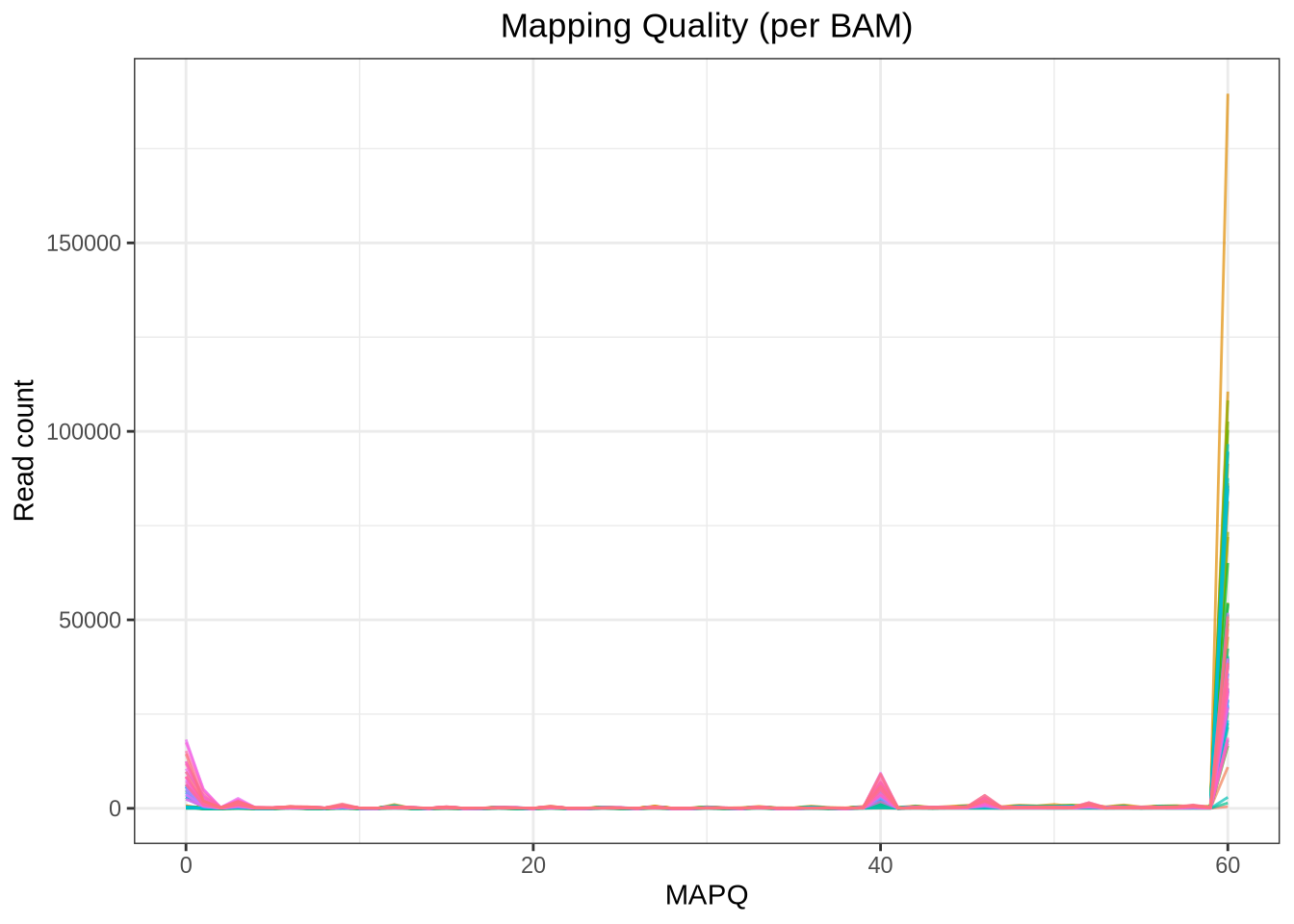
Plot: Mapping Quality Distribution
Code
# Load required packagessuppressPackageStartupMessages({library(Rsamtools)library(ggplot2)library(dplyr)})# Set working directory to BAM foldersetwd("/home/tahirali/RAMSES_mount/tali/qbio/Practical_Day_1/mapping")# Get list of all BAM files in the directorybam_files <-list.files(pattern ="\\.bam$", full.names =TRUE)# Function to extract MAPQ from BAMget_mapq <-function(bamfile) { param <-ScanBamParam(what ="mapq") aln <-scanBam(bamfile, param = param)[[1]]data.frame(MAPQ = aln$mapq, Sample =basename(bamfile))}# Extract MAPQ for all BAMsmapq_list <-lapply(bam_files, get_mapq)mapq_df <-bind_rows(mapq_list)# Clean: remove NAmapq_df <- mapq_df %>%filter(!is.na(MAPQ), MAPQ >=0)# Density plot across BAMsggplot(mapq_df, aes(x = MAPQ, color = Sample)) +geom_density(size =0.7, alpha =0.5) +theme_bw() +theme(plot.title =element_text(hjust =0.5), # center titlelegend.position ="none" ) +labs(title ="Mapping Quality Distribution across BAMs",x ="MAPQ score", y ="Density" )
Code
# line plot of MAPQ counts per BAMmapq_summary <- mapq_df %>%group_by(Sample, MAPQ) %>%summarise(Count =n(), .groups ="drop")ggplot(mapq_summary, aes(x = MAPQ, y = Count, color = Sample)) +geom_line(alpha =0.7) +theme_bw() +theme(plot.title =element_text(hjust =0.5), # center titlelegend.position ="none" ) +labs(title ="Mapping Quality (per BAM)",x ="MAPQ", y ="Read count" )
my_lib <-"/run/media/tahirali/2a4efc98-aaa3-4a01-a063-37db1463f3a8/tools/anaconda/envs/quarto-env/lib/R/library"cran_pkgs <-c("optparse", "XML")bioc_pkgs <-c("NOISeq", "Rsamtools", "Repitools", "rtracklayer")is_installed <-function(pkg, lib) { pkg %in%rownames(installed.packages(lib.loc = lib))}# Set CRAN mirror only for CRAN packagesoptions(repos =c(CRAN ="https://cloud.r-project.org"))for (pkg in cran_pkgs) {if (!is_installed(pkg, my_lib)) {install.packages(pkg, lib = my_lib) }}# Do NOT set options(repos=...) before BiocManager::install()if (!is_installed("BiocManager", my_lib)) {install.packages("BiocManager", lib = my_lib)}BiocManager::install(version ="3.20")for (pkg in bioc_pkgs) {if (!is_installed(pkg, my_lib)) { BiocManager::install(pkg, lib = my_lib) }}
Example SLURM Script for BAMQC Analysis
#!/bin/bash -l#SBATCH --job-name=bamqc#SBATCH --time=1:00:00#SBATCH --cpus-per-task=4#SBATCH --mem=16gb#SBATCH --account=UniKoeln#SBATCH --output=/home/group.kurse/qbius030/Practical_Day_1/bamqc/bamqc.out#SBATCH --error=/home/group.kurse/qbius030/Practical_Day_1/bamqc/bamqc.errBAM_DIR="/home/group.kurse/qbius030/Practical_Day_1/mapping"BAMQC_DIR="/home/group.kurse/qbius030/Practical_Day_1/bamqc"MULTIBAMQC_DIR="$BAMQC_DIR/multibamqc"mkdir-p"$BAMQC_DIR""$MULTIBAMQC_DIR"# Run Qualimap BAMQC for each sorted BAM filefor BAM_FILE in"$BAM_DIR"/*.sorted.bam;doSAMPLE_NAME=$(basename"$BAM_FILE" .sorted.bam)QUALIMAP_OUTPUT_DIR="$BAMQC_DIR/$SAMPLE_NAME"echo"Running Qualimap BAMQC for $SAMPLE_NAME"qualimap bamqc \-bam"$BAM_FILE"\-outdir"$QUALIMAP_OUTPUT_DIR"\-outformat HTML \-outfile"${SAMPLE_NAME}_report"done# Prepare input groups file for multi-bamqcINPUT_GROUPS_FILE="$BAMQC_DIR/input_groups.txt"rm-f"$INPUT_GROUPS_FILE"echo"Creating input groups file for Multi-BAMQC"for BAMQC_OUTPUT_DIR in"$BAMQC_DIR"/*;doif[-d"$BAMQC_OUTPUT_DIR"]&&[-f"$BAMQC_OUTPUT_DIR/genome_results.txt"];thenSAMPLE_NAME=$(basename"$BAMQC_OUTPUT_DIR")echo-e"${SAMPLE_NAME}\t${BAMQC_OUTPUT_DIR}\t1">>"$INPUT_GROUPS_FILE"fidone# Run Qualimap Multi-BAMQCecho"Running Qualimap Multi-BAMQC"qualimap multi-bamqc \-d"$INPUT_GROUPS_FILE"\-outdir"$MULTIBAMQC_DIR"\-outformat PDFecho"Qualimap analysis complete. Reports saved in $MULTIBAMQC_DIR."
View results using Firefox:
cd /home/group.kurse/qbius030/Practical_Day_1/bamqc/multibamqcfirefox report.pdf
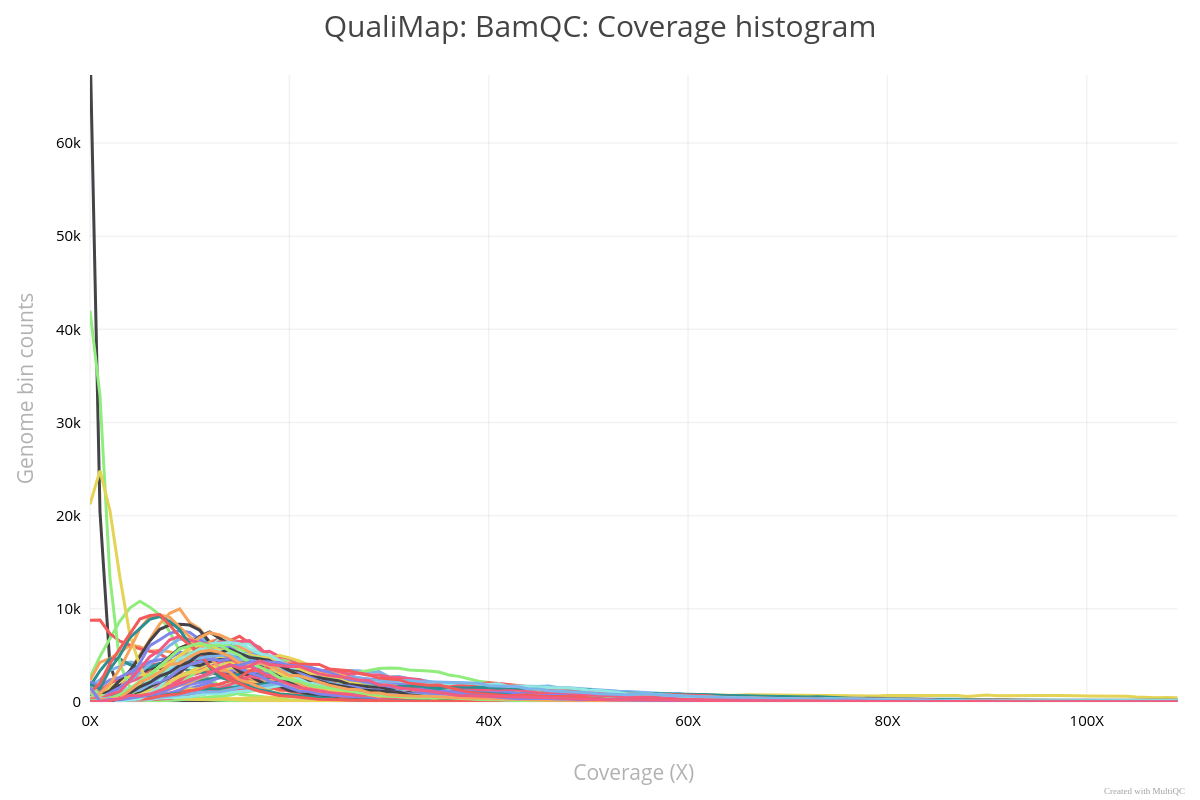
Use MultiQC to Summarize Qualimap QC of All Samples
Thank you for your active participation today! 🎉
Great job on tackling today’s challenges—we hope you found it engaging and insightful.
See you next week for more exciting learning and discoveries! Keep practicing and exploring until then! 🚀
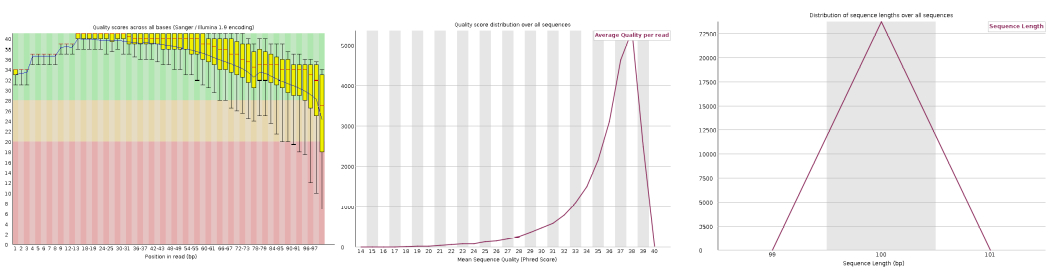 Figure: FastQC per-base quality plot
Figure: FastQC per-base quality plot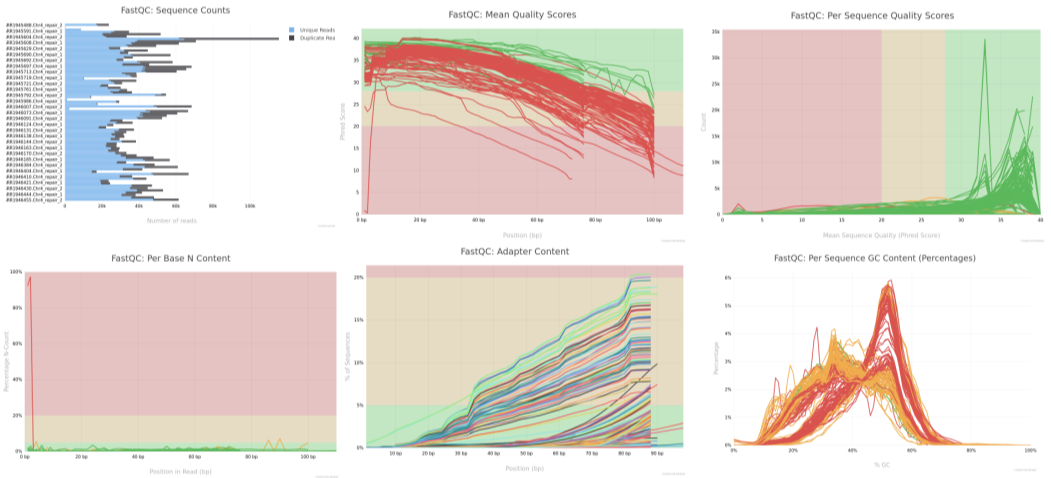 Figure: MultiQC Summary (after running MultiQC on FastQC results)
Figure: MultiQC Summary (after running MultiQC on FastQC results)